NOC Tutorials
- Installation and getting start
- For Protein structure visualizing, analyzing
- For general crystallographic protein structure determination purpose
- For GROMACS, AMBER MD trajectories displaying
- A simple development guide
Installation and Getting Start
NOC is open source code and currently precompiled binaries are available for MS Windows, Linux, Mac OSX, FreeBSD, Solaries.
We recommend you to build your machine's binary from the source code for Linux/Unix platform.Precompiled binary installation
A precompiled binary installation is very easy, downloading NOC from NOC downloading page,for MS-Windows, you only need download noc.zip, unpack it and launch noc.exe;
for Mac OSX, you need download "noc.osx.ppc.zip" and nocdat.zip, unpack them, move nocdat to your home directory, and launch noc.app;
for Linux, you need download nocdat.tar.gz and noc.fc4.gz, unpack nocdat.tar.gz by command tar xzf nocdat.tar.gz and move nocdat to your home directory, unpack noc.fc4.gz by command gunzip noc.fc4.gz and rename it to noc, move it your executable or make a link, type noc to launch it;
for FreeBSD, following above but download noc.bsd62.gz rather than noc.fc4.gz
for Solaris, following above but download noc.sol10.gz rather than noc.fc4.gzBuilding from the source code:
1. Go to NOC downloading page, download noc_src.tar.gz to your home directory and unpack it, download wxWindows wxGTK-2.6.3.patched.multisampling.tgz for Linux/Unix, wxMac-2.6.3.multisampling.tgz for MacOSX or get it from http://www.wxwidgets.org. Unpack wxWidgets and type "sh autoconfigure.sh" to configure it, make to compile it, after make copy wx-config to NOC source code folder. Note: If you don't have GTK2, please configure with "--with-gtk=1". Only wxWidgets2.6.3 is fully tested with current NOC source code, but the higher version should work too. The wxWidgets packages provided by us support OpenGL Multisample Antialiasing, which is available for most of recent graphic adapter and make real-time visualizing better.
2. In NOC source code directory, type command "sh autolibsetup.sh" to compile libraries depended. "sh compile.sh" to compile NOC. Notes: You can find the original libraries in http://noch.sourceforge.net/#Related_Links if you want to build newer version libraries for NOC. The final output is a file named "noc", copy it to your executable path. A app bundle noc.app will be generated for MacOSX building.
3. If you don't have "nocdat.tar.gz" installation yet, please download it and unpack it to your home directory first.
Notes:
1. It's better to have #version of GTK with development library > 2.0
2. For some Unix(like FreeBSD, Solaris), #version of bash > 2.05 is needed to build FFMpeg librariy.NOC can be run both in GUI mode and in command line mode (You may want to run NOC for some batch job, to run NOC in command mode by noc x or rename noc to nocconsole).
Usage: noc [options] [files] [@NOC internal commands]
Options:-h --help #show this help message and exit
-c --curpath #reset all work directories to current dir
-r --reset #reset all NOC profile to default value
-a --amber #load AMBER trajectories, see example
-g --gmx #load GROMACS gro file, see example
-s --stereo #start NOC with hardware stereo support
-x --stereo #start NOC in console mode, i.e. without GUI
-uma #disable Multisampling
-v --version #show versionnoc 1pgb.pdb
#load a pdb filenoc -a tc5b.top tc5b.md.x
#load AMBER top and trj filenoc -g 2spz.gro [2spz.md.xtc|.trr]
#load GROMACS gro and xtc or trrnoc -x pdb1gpb.pdb @distance a.16.SG a.30.SG @exit s
#run NOC in command mode, load structure, compute the distance between residue #16 SG and residue #30 SG, and quit NOCnoc -x pdb1gpb.pdb @dihe a.16.CB a.16.SG a.30.SG a.30.CB @exit s
#run NOC in command mode, load structure, compute the dihedirals CB-SG-SG-CB and quit NOCnoc -x @script caldihe.txt
#run NOC in command mode and load script caldihe.txt
caldihe.txt content is:
readpdb pdb1gpb.pdb
dihe a.16.CB a.16.SG a.30.SG a.30.CB
exit -sNote: most of NOC functions are available by mouse clicking in GUI mode, the command line mode is only for special purpose.
For Protein structure visualizing, analyzing
Loading a structure
Structural representation and coloring
Mouse and keyboard operating
Make a movie and draw to image file
Molecular electronstatic/hydrophobic surface
Structure analysis
Loading a molecule
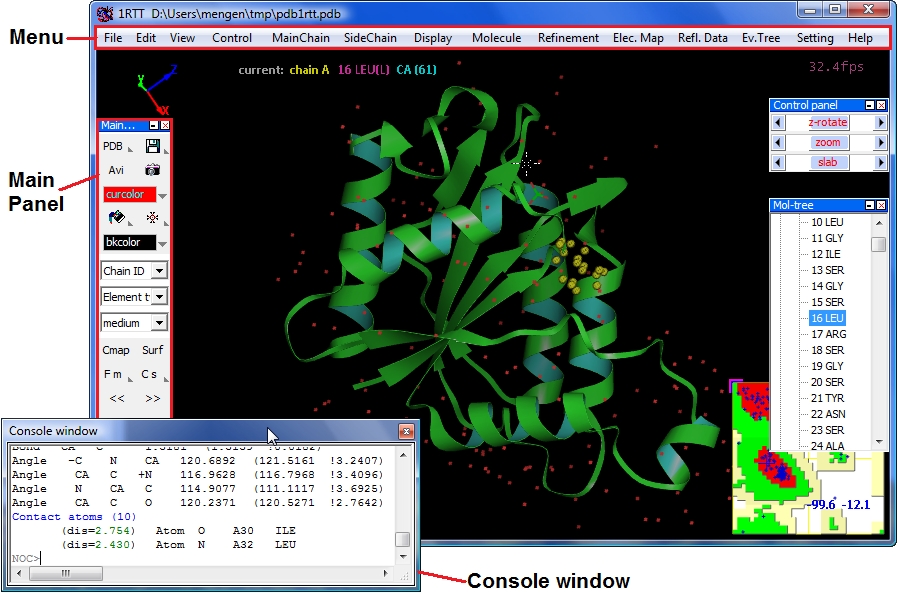
Menu --> "File" --> "Open molecule", currently NOC only support PDB format structure data, and actually NOC can read any particle system in PDB format.
or Menu --> "Fetch from RCSB PDB" to download molecular data from RCSB Protein Data Bank by PDB ID like "1RTT"
or Input a molecular data file as a argument when launching NOC like "noc pdb1rtt.pdb"When loading a molecule, NOC will do automatically structural checking, residue/atom nomenclature checking, missing atoms adding/fitting, hydrogen atoms adding/fitting, secondary structure assignment, structural topology building, basic parameters assignment, and so on.
Note: NOC supports reading multi-models as trajectories, which will enable the trajectories panel to help do some structure analysis. Currently, NOC only supports one molecule for one instance, but you can load new molecule as "appending to current molecule".
Structural representation and coloring
MainChain representation:
Menu --> "MainChain"
SideChain representation:
Menu --> "SideChain"
Hetgroup representation:
Menu --> "Display"-->"Hetgroup in CPK/Ware/Stick"
Water representation:
Menu --> "Display"-->"Water in Lines/Stick/Ball"
MainChain coloring themes:
Menu --> "MainChain" --> "Color theme by" --> "Index"
SideChain coloring themes:
Menu --> "SideChain" --> "Color theme by" --> "Element type"
mouse Left button down on a object to pick it (+ Shift down for multi-selecting)
mouse Left button down + move to rotate scene in XY plane
mouse Left button down + move + shift to translate in XY plane
mouse Left button double click on a object to label it, on empty to release selections
mouse wheel up/down to zoom in/outkeyboard "C" to move center to current atoms selected
keyboard Shift + "C" to move center to molecular centroid
keyboard "N" to move center to next residue
keyboard "B" to move center to back residueNOC supports auto spinning: Menu --> "Control" --> "Auto spin" to Enable/Disable
Make a movie and export to graphic
Menu --> "File" --> "Make move" to make a short movie
Menu --> "File" --> "Draw to image file" to export scene to a graphic file (supports BMP/JPEG/PNG/GIF/TIFF format)
Menu --> "File" --> "Draw to ps file" to make high quality PostScript print, but currently it works slowly on MS Windows.
Menu --> "File" --> "Copy screen" to make a scene shoot
Molecular surface representation
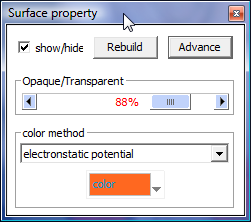
to build a molecular surface: Menu --> "Display" --> "Solid surface (Build)" to show a interactive dialog to help you compute molecular surface
after surface building, it will show "Surface property" panel, you can change color method and opaque here, try more functions in "Advance" button
to change the electronstatic potential or hydrophobic force coloring scaling by Shift + Mouse left button down in the Color meter + Move Down/Up.
to change sites definition and potential/force function, please read the code: "function CalculateElectricPotential()/CalculateHyrophobicForce()" in file "src/solidsurface.cpp".
Instant picking note
when picking a atom, NOC will show all bonds, angles, dihedrals information (include structural error) involving the atom instantly. For example, when picking a CYX "SG" atom it will show you the dihedral angle of S-S_BOND "CB-S-S-CB".Measure distance, angle, and dihedral by commands in NOC Console Window:
distance a.2.CA a.10.CA #report distance between CA atom of Residue #2 of Chain A and CA atom of Residue #10 of Residue #10 of Chain A
distance a.2.CA a.10.CA a.20.CA #report angle between CA atom of Residue #2 of Chain A...
dihe a.2.CA a.10.CA a.20.CA a.20.N #report dihedrals between CA atom of Residue #2 of Chain A...Structure fitting/comparison
to compare two chains: Menu --> "Molecule" --> "Compare chains", it will show a dialog to guide you do structures fitting including sequence alignment and reports the difference in RMSD.
Note: if you want compare two molecules, please try merging molecules firstCalculate molecular solvent accessible surface and buried/contacting surface:
Menu --> "Molecule" --> "Calculate accessible surface in details", it will report whole molecular solvent accessible surface (SAS), individual residue's SAS, individual chain's SAS, plotting residues SAS, and buried/contacting surface among chains.Ramachandran plot/MainChain dihedrals
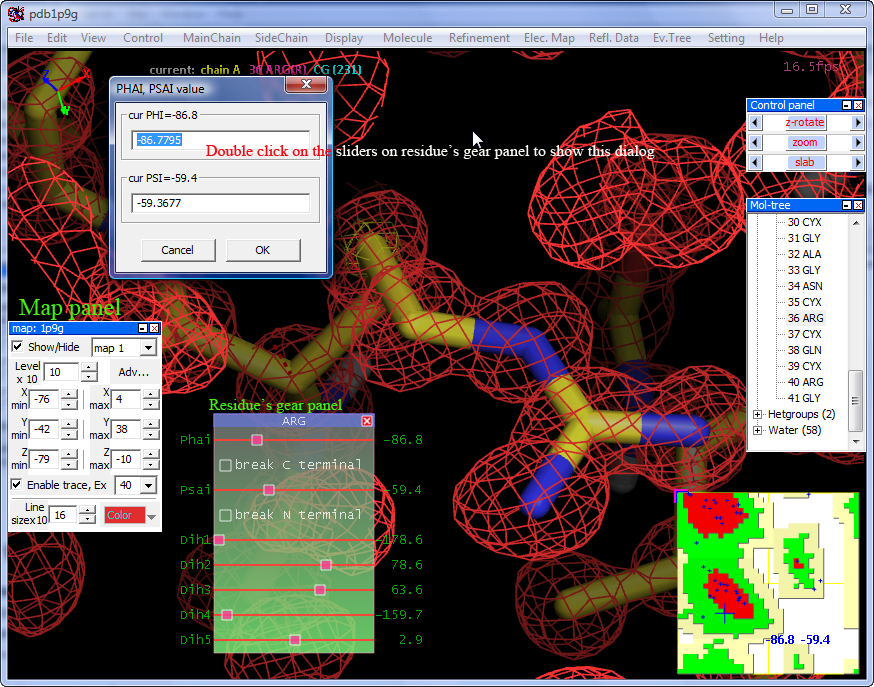
Menu --> "Display" --> "Rama. Plot" to show Ramachandran plot, and on Ramachandran plot "Right mouse button down" to show a popup menu...
"Report details" to report all residues' PHI and PSI angles
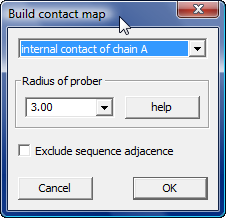
Contact maps
Menu --> "Display" --> "Contacts map (Build)" to show a dialog to help build contact map
Secondary structures
Menu --> "Molecule" --> "Assign/reassign secondary structures" to reassign secondary structure types in details in NOC Console Window
Note: when loading molecular data, NOC already assigned secondary structures by not printing the details.More structural analysis functions please try Menu --> "Molecule"
For general crystallographic protein structure determination purpose
Basic crystallographic functions for structure analysis
Synthesis a electron density map from HKL data (indexed diffraction data)
Loading a electron density map created by CNS/CCP4
Structure gearing/modeling
Structure refinementBasic crystallographic functions for structure analysis
To do a crystallographic analysis, you must have your molecular data with valid unit cell parameter and space group.
to Show periodic cell box: Menu --> "Display" --> "Show unit cell frame"
to Show symmestry related molecules: Menu --> "Molecule" --> "Build related molecules (by Cryst. Sym.)"
to Show contact molecules in crystal: Menu --> "Molecule" --> "Build contact molecules"
to Show crystal packing: Menu --> "Molecule" --> "Draw crystal packing"
try Menu --> "Molecule" --> "Advance" for more symmestry related translation/rotationNote: Cooperating with the basic structural analysis functions can help you to valid the structure.
Synthesis a electron density map from HKL data
To synthesis a electron density map, you need:
1. Load a structural model and an indexed diffraction data
Menu --> "File" --> "Open HKL(reflection) data" to read HKL file, current NOC support MTZ(CCP4),CIF(PDB),CV(CNS,XPLOR) format
2. Menu --> "Refl. Data" --> "Calculate structure factor" to compute the structural phase
3. Menu --> "Refl. Data" --> "Synthesis electron density map" to show a dialog to guide you to synthesis a map
Loading a electron density map created by CNS/CCP4
to loading electron density map: Menu --> "File" --> "Open Electron Density Map",
Currently NOC supports CCP4, XPLOR/CNS, DSN6, BRIX(O) format, but you must load a molecular model first.
After map loaded successful it will show the "Map panel ", it's no harm to try the controls on the panel.
Grabbing/moving atoms
selecting atoms/groups by mouse left button click (+Shift down for multi-selection)
Ctrl + mouse move to move the atoms selected in XY plane
Ctrl + Shift + mouse move to move in Z directionResidue's dihedrals gear panel:
Menu --> "View" --> "Dihedrals panel" to enable residue's dihedrals gear panel
Structure refinement
Currently, NOC uses Amber 2003 full atoms forcefield for local/global structure energy minimization.
to do a local energy minimization, you need selection atoms/groups first,
then go Menu --> "Refinement" --> "Fit atoms selected(All other atoms will be fixed)"Global energy minimization: Menu --> "Refinement" --> "Overall Energy minimize"
Note: we recommend you do a local energy minimizing for each local structure modification.
For GROMACS, AMBER MD trajectories displaying
NOC supports GROMACS, AMBER format trajectories, and NOC will keep your original MD data untouched.
to load Gromacs MD trajectories : Menu --> "Open Gromacs files", or launching NOC by "noc -g <tpr/gro file> <trr/xtc file>"
to load AMBER MD trajectories: Menu --> "Open Amber files", or launching NOC by"noc -a <top/pdb file> <crd file>"
There are many functions for trajectories analysis, help yourself.
NOC is coded in C++, the code structures is simple and extendable. Following is a example how to implement a internal command in NOC.
1. open "src/molecule.h" to declare a function:
void CalculateMolWeight_test(bool bhet, bool bwat);
open "src/molecule.cpp" to implement the function definition:
float CMolecule::CalculateMolWeight_test(bool bhet, bool bwat)
{
register float molWeight=0.0f;
EACH_RESIDUE_IN_MOL;
{
molWeight +=Chain[nC]->residue[nR]->getWeight();
}
L_LOOP;
if(bhet)
{
for(nR=0; nR<this->hetChains.GetNumGroups(); nR++)
molWeight +=hetChains.groups[nR].getWeight();
}if(bwat)
{
for(nR=0; nR<numWat; nR++)
molWeight +=wat[nR].getWeight();
}
return molWeight;
} //you might need to read the CMolecule, CChain, CResidue, CAtom class structure first2. open "src/noccmd.cpp" to add a command:
in "void CNOCCmd::_addCmdList()" function insert a line:
sCmdList.Add("calc_molweight_test");
in "int CNOCCmd::_execute()" function insert:
else if(isCommand("cal_molweight_test"))
{
if(g_pMol==NULL){print("\nplease load molecule first"); return nocCMD_SUCCESS;}
print("\nMolecule weight="); print(g_pMol->CalculateMolWeight_test(false, false));
}3. Recompiling NOC by command "make" and run a new NOC instance, load a protein and in NOC Console Winodw type "calc_molweight_test" to test the command.